|
|
|
Previous | Next
doi:10.2204/iodp.proc.311.208.2008
X-ray diffraction methods
Sample preparation
The sample preparation method used in this study is described in Holtzapffel (1985). Separation of clay-sized fractions (<2 μm started with a gentle crushing of samples and decalcification using a solution of 0.2N HCl in an Erlenmeyer flask. The preparation was washed with distilled water, and clay deflocculation was obtained through repeated centrifuging (2500 revolutions per minute [rpm] for 3 min). After each centrifugation, water was eliminated and the plug was resuspended in distilled water. Generally, three to six centrifugation–suspension cycles were necessary until deflocculation. After transferring the suspended sediment to a glass beaker and vigorously shaking it, the clay fraction was separated by decantation using settling time based on Stoke's law. The extracted clay fraction was then centrifuged at 3500 rpm for 40 min, and, finally, the clay plug was used to make an oriented clay glass slide. X-ray diffraction analyses were conducted on air-dried clay slides after saturation with ethylene glycol (at least 24 h) and after heating at 490°C for 2 h.
X-ray diffraction parameters
X-ray diffractograms were obtained at the Muséum National d'Histoire Naturelle of Paris using a Siemens D500 X-ray diffractometer with CuK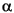 radiation (1.54 Å) and Ni filter. Instrument parameters were set to 40 kV accelerating voltage and 30 mA current. Scans were run from 2° to 40°2 radiation (1.54 Å) and Ni filter. Instrument parameters were set to 40 kV accelerating voltage and 30 mA current. Scans were run from 2° to 40°2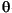 with a scanning step size of 0.02°2 and counting time of 4 s. X-ray diagrams were studied using MacDiff (version 4.2.5) software (servermac.geologie. with a scanning step size of 0.02°2 and counting time of 4 s. X-ray diagrams were studied using MacDiff (version 4.2.5) software (servermac.geologie.
uni-frankfurt.de/Staff/Homepages/Petschick/RainerE.html) in order to establish the background line, smooth counts, correct peak positions (using quartz [100] peak at 4.24 Å), and compute integrated peak areas (total counts). The semiquantitative clay mineral proportions were estimated from the glycolated pattern. Clay mineral identification was made according to the position of the 001 series of basal reflections (Brown and Brindley, 1980). X-ray diffraction analyses allowed us to identify smectite, illite, chlorite, kaolinite, and some random mixed-layered illite/vermiculite (I/V). These I/V mixed layers are characterized in air-dried conditions by a broad peak ranging from 10 to 13 Å, with a maximum intensity at ~12 Å. This peak is not altered in position or intensity by glycol saturation but collapses to 10 Å after heating (Fig. F2). In order to allow comparisons with published data from the northern Pacific region (Duncan et al., 1970; Karlin, 1980; Underwood, 2002; Underwood and Torres, 2006), the integrated areas were multiplied by weighting factors (Biscaye, 1965) and normalized to 100%. Weighting factors are 4 for illite, 2 for kaolinite + chlorite, and 1 for smectite (Biscay, 1965); we chose to use 1 for I/V in order to compare results with those already published for this area (in which no I/V was detected), so percentages are only slightly "mathematically" modified. Chlorite and kaolinite were differentiated using the 3.54 Å/3.57 Å peak ratio. The smectite term corresponds to a random R0 smectite/illite mixed layer (R0 I/S) and the amount of expandable layers was calculated based on the valley/peak height (v/p) ratio (after Biscay, 1965), the difference in the height of the base line from the high and low 2 sides of the 5.2°2 peak. Abacus allows consideration of the percentage of smectite portion from the measure of the v/p parameter (the saddle-peak method; Rettke, 1981). The estimated analytical precision is ±5%.
Top of page | Previous | Next |

